Kawasaki Disease

Kawasaki disease is a syndrome of unknown cause that results in a fever and mainly affects children under 5 years of
age. It is a form of vasculitis, where blood vessels become inflamed throughout the
body. The fever
typically lasts for more than five days and is not affected by usual medications. Other common symptoms include large lymph nodes in the neck, a rash in the genital area, lips, palms, or soles of the feet, and red eyes. Within three
weeks of the onset, the skin from the hands and feet may peel, after which
recovery typically occurs. In some children, coronary artery aneurysms form in the heart.
While the specific cause is unknown, it is thought to
result from an excessive immune system response to an infection in children who are genetically predisposed. It does not spread between people. Diagnosis is
usually based on a person's signs and symptoms. Other tests such as an ultrasound of the heart and blood tests may support the diagnosis. Diagnosis
must take into account many other conditions that may present similar features,
including scarlet fever and juvenile rheumatoid arthritis. An emerging Kawasaki-like' disease temporally associated with COVID-19 appears to be a distinct syndrome.
Typically, initial treatment of Kawasaki disease consists of high doses of aspirin and immunoglobulin. Usually, with treatment, fever resolves within 24 hours and full recovery occurs. If the coronary arteries are involved, ongoing treatment or surgery may occasionally be required. Without treatment, coronary artery aneurysms occur in up to 25% and about 1% die. With treatment, the risk of death is reduced to 0.17%. People who have had coronary artery aneurysms after Kawasaki disease require lifelong cardiological monitoring by specialized teams.
Kawasaki disease is rare. It affects between 8 and 67 per 100,000 people
under the age of five except in Japan, where it affects 124 per 100,000. Boys are more commonly affected than girls. The disorder is
named after Japanese pediatrician Tomisaku Kawasaki, who first described it in 1967.
Signs and symptoms

Signs of Kawasaki
disease
Kawasaki disease often begins with a high and persistent
fever that is not very responsive to normal treatment with paracetamol acetaminophen) or ibuprofen. This is the most prominent symptom of Kawasaki
disease, and is a characteristic sign that the disease is in its acute phase;
the fever normally presents as a high (above 39–40 °C) and remittent, and is followed by extreme irritability. Recently, it
is reported to be present in patients with atypical or incomplete Kawasaki
disease; nevertheless, it is not present in 100% of cases.
The first day of fever is considered the first day of the
illness,
and its duration is
typically one to two weeks; in the absence of treatment, it may extend for
three to four weeks. Prolonged fever is associated with a higher incidence of cardiac
involvement. It responds partially
to antipyretic drugs and does not cease with the introduction of antibiotics. However, when appropriate therapy is
started – intravenous immunoglobulin and aspirin the fever subsides after two days.
Bilateral conjunctival inflammation has been reported to be the most common symptom
after fever.
It typically
involves the bulbar conjunctivae, is not accompanied by suppuration, and is not
painful. This usually begins shortly after the onset of fever during the
acute stage of the disease. Anterior uveitis may be present under slit-lamp examination. Iritis can occur, too. Keratic precipitates are another eye manifestation detectable by a slit
lamp but are usually too small to be seen by the unaided eye.
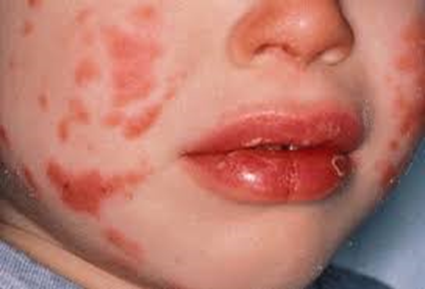
Kawasaki disease also presents with a set of mouth
symptoms, the most characteristic of which are a red tongue, swollen lips with
vertical cracking, and bleeding. The mucosa of the mouth and throat may be bright red, and the tongue may have a typical "strawberry tongue" appearance (marked redness with prominent gustative
papillae. These mouth symptoms
are caused by necrotizing microvasculitis with fibrinoid
necrosis.
Cervical lymphadenopathy is seen in 50% to 75% of children, whereas the
other features are estimated to occur in 90%, but sometimes it can be the dominant presenting
symptom. According to the diagnostic criteria, at least one
impaired lymph node ≥ 15 mm in diameter should be involved. Affected lymph nodes are painless or minimally
painful, nonfluctuant, and nonsuppurative; erythema of the neighboring skin may
occur. Children with fever and neck adenitis who do not respond to antibiotics should have
Kawasaki disease considered as part of the differential diagnoses.
|
Less common manifestations |
|
|
System |
Manifestations |
|
Diarrhea, chest pain, abdominal
pain, vomiting, liver dysfunction, pancreatitis, hydrops gallbladder, parotitis, cholangitis, intussusception, intestinal pseudo-obstruction, ascites, splenic infarction |
|
|
Myocarditis, pericarditis, tachycardia, valvular heart disease |
|
|
Urethritis, prostatitis, cystitis, priapism, interstitial nephritis, orchitis, nephrotic syndrome |
|
|
Lethargy, semicoma, aseptic meningitis,
and sensorineural deafness |
|
|
Shortness of breath, influenza-like illness, pleural
effusion, atelectasis |
|
|
Skin |
Erythema and induration at BCG vaccination site, Beau's lines, and finger gangrene |
|
Source: review, table. |
|
In the acute phase of the disease, changes in the peripheral extremities can include erythema of the palms and soles, which is often striking with sharp demarcations and
often accompanied by painful, brawny edema of the dorsa of the hands or feet,
so affected children frequently refuse to hold objects in their hands or to
bear weight on their feet. Later, during the convalescent or the subacute
phase, desquamation of the fingers and toes usually begins in the periungual region within two to three weeks after the onset of fever
and may extend to include the palms and soles. Around 11% of children affected
by the disease may continue skin-peeling for many years. One to two months
after the onset of fever, deep transverse grooves across the nails may develop
(Beau's lines), and occasionally nails are shed.
The most common skin manifestation is a diffuse macular-papular erythematous rash, which is quite nonspecific. The rash varies over time and is characteristically
located on the trunk; it may further spread to involve the face, extremities,
and perineum. Many other forms of cutaneous lesions have been
reported; they may include scarlatiniform, papular, urticariform, multiform-like erythema, and purpuric lesions; even micropustules were reported. It can be polymorphic, not itchy, and normally observed up to the fifth day of
fever. However, it is never bullous or vesicular.
In the acute stage of Kawasaki disease, systemic
inflammatory changes are evident in many organs. Joint pain arthralgia) and swelling,
frequently symmetrical, and arthritis can also occur. Myocarditis, diarrhea, pericarditis, valvulitis, aseptic
meningitis, pneumonitis, lymphadenitis, and hepatitis may be present and are manifested by the presence
of inflammatory cells in the affected tissues. ] If left untreated, some symptoms will eventually
relent, but coronary artery aneurysms will not improve, resulting in a
significant risk of death or disability due to myocardial infarction. If treated quickly, this risk can be mostly
avoided and the course of illness cut short.

Signs and symptoms
and time course of Kawasaki disease
Other reported nonspecific symptoms include cough, rhinorrhea, sputum, vomiting, headache, and seizure.
The course of the disease can be divided into three
clinical phases.
·
The acute febrile phase, which usually lasts for one to two
weeks, is characterized by fever, conjunctival injection, erythema of the oral mucosa, erythema and swelling of
the hands and feet, rash, cervical adenopathy, aseptic
meningitis, diarrhea, and hepatic dysfunction. Myocarditis is common during this time, and a pericardial effusion
may be present. Coronary arteritis may be present, but aneurysms are generally not yet visible by echocardiography.
·
The subacute phase begins when fever, rash, and lymphadenopathy resolve at about one to two weeks after the onset
of fever, but irritability, anorexia, and conjunctival injection persist.
Desquamation of the fingers and toes and thrombocytosis are seen during this stage, which generally lasts
until about four weeks after the onset of fever. Coronary artery aneurysms usually develop during this time, and the
risk for sudden death is highest.
·
The convalescent stage begins when all clinical signs of illness
have disappeared, and continues until the sedimentation rate returns to normal, usually at six to eight weeks
after the onset of illness.
Adult onset of Kawasaki disease is rare. The presentation differs between adults and
children: in particular, it seems that adults more often have cervical
lymphadenopathy, hepatitis, and arthralgia.
Some children, especially young infants, have atypical presentations without the classic
set of symptoms Such presentations are associated with a higher risk of
cardiac artery aneurysms.
Cardiac

X-ray showing
aneurysmal enlargement of the coronary arteries, which is a complication in a
Kawasaki syndrome.
Heart complications are the most important aspect of
Kawasaki disease, which is the leading cause of heart disease acquired in
childhood in the United States and Japan. In developed nations, it appears
to have replaced acute rheumatic fever as the most common cause of acquired heart disease
in children. Coronary artery aneurysms occur as a sequela of the
vasculitis in 20–25% of untreated children. It is first detected at a mean of 10 days of
illness and the peak frequency of coronary artery dilation or aneurysms occurs
within four weeks of onset. Aneurysms are classified into small (internal
diameter of vessel wall <5 mm), medium (diameter ranging from
5–8 mm), and giant (diameter > 8 mm). Saccular and fusiform aneurysms usually develop
between 18 and 25 days after the onset of illness.
Even when treated with high-dose IVIG regimens within the first 10 days of illness, 5% of
children with Kawasaki disease develop at the least transient coronary artery
dilation and 1% develop giant aneurysms. Death can occur
either due to myocardial infarction secondary to blood clot formation in a coronary artery aneurysm or to rupture of a large coronary artery aneurysm. Death is most
common two to 12 weeks after the onset of illness.
Many risk factors predicting coronary artery aneurysms
have been identified, including persistent fever after IVIG
therapy, low hemoglobin concentrations, low albumin concentrations, high white-blood-cell count, high band count, high CRP concentrations, male sex, and age less than one
year. Coronary artery lesions resulting from Kawasaki disease change
dynamically with time. Resolution one to two years after the onset of the
disease has been observed in half of vessels with coronary aneurysms. Narrowing of the coronary artery, which occurs as a result of
the healing process of the vessel wall, often leads to significant obstruction
of the blood vessel and the heart not receiving enough
blood and oxygen. This can eventually lead to heart muscle tissue
death, i.e., myocardial infarction (MI).
MI caused by thrombotic occlusion in an aneurysmal,
stenotic, or both aneurysmal and stenotic coronary artery is the main cause of
death from Kawasaki disease.] The highest risk of MI occurs in the first year
after the onset of the disease. MI in children presents with different symptoms
from those in adults. The main symptoms were shock, unrest, vomiting, and abdominal pain; chest pain was most common in older children. Most of these children had the attack occurring
during sleep or at rest, and around one-third of attacks were asymptomatic.
Valvular insufficiencies, particularly of mitral or tricuspid valves, are often observed in the acute phase of Kawasaki
disease due to inflammation of the heart valve or inflammation of the heart muscle-induced myocardial dysfunction, regardless of coronary
involvement. These lesions mostly disappear with the resolution of acute
illness, but a very small group of the lesions persist and progress. There
is also late-onset aortic or mitral insufficiency caused by thickening or deformation of fibrosed valves, with the timing ranging from several months to years
after the onset of Kawasaki disease. Some of these lesions require valve replacement.
Other
Other Kawasaki disease complications have been described,
such as aneurysm of other arteries: aortic aneurysm, with a higher number of reported cases involving
the abdominal aorta, axillary artery aneurysms brachiocephalic artery aneurysm, ] aneurysm of iliac and femoral arteries, and renal artery aneurysm. Other vascular complications can occur such as
increased wall thickness and decreased distensibility of carotid arteries, and brachioradial artery. This change in the vascular tone is secondary to
endothelial dysfunction. In addition, children with Kawasaki disease, with
or without coronary artery complications, may have a more adverse
cardiovascular risk profile, such as high blood pressure, obesity, and abnormal serum lipid profile
Gastrointestinal complications in Kawasaki disease are
similar to those observed in Henoch–Schönlein purpura, such as: intestinal obstruction, colon swelling, intestinal ischemia, intestinal pseudo-obstruction, and acute abdomen.
Eye changes associated with the disease have been
described since the 1980s, being found as uveitis, iridocyclitis, conjunctival hemorrhage, optic neuritis, amaurosis, and ocular artery obstruction. It can also be found as necrotizing vasculitis,
progressing into peripheral gangrene.
The neurological complications per central nervous system
lesions are increasingly reported. The neurological complications found are meningoencephalitis, subdural
effusion, cerebral hypoperfusion, cerebral
ischemia and infarct,[ cerebellar infarction, manifesting with seizures, chorea, hemiplemental confusion, lethargy and coma, or even a
cerebral infarction with no neurological manifestations. Other neurological
complications from cranial nerve involvement
are reported as ataxia, facial palsy, and sensorineural hearing lossgia .Behavioral changes are thought to be caused by localized cerebral hypoperfusion, can include attention deficits, learning deficits,
emotional disorders emotional
lability, fear of night, and night terrors), and internalization problems (anxious, depressive or aggressive behavior).
Causes
The specific cause of Kawasaki disease is unknown.
A plausible
explanation is that it may be caused by an infection that triggers an inappropriate immunologic cascade in a small number of genetically predisposed children. The pathogenesis is complex and
incompletely understood. Various
explanations exist.
Circumstantial evidence points to an infectious cause. Since recurrences are unusual in Kawasaki disease, it is thought that
the trigger is more likely to be represented by a single pathogen, rather than a range of viral or bacterial
agents. Various candidates have been implicated, including upper respiratory tract infection by some novel RNA virus. Despite intensive search, no single pathogen has
been identified. There has been debate as to whether the infectious agent
might be a superantigen (i.e. one commonly associated with excessive immune
system activation). Current
consensus favors an excessive immunologic response to a conventional antigen
which usually provides future protection. Research points to nunidentified ubiquitous virus, possibly one that enters through the respiratory
tract.
Seasonal
trends in the appearance of new cases of Kawasaki disease have been linked
to tropospheric wind patterns, which suggests wind-borne transport
of something capable of triggering an immunologic cascade when inhaled by
genetically susceptible children. Winds blowing from central Asia correlate with
numbers of new cases of Kawasaki disease in Japan, Hawaii, and San Diego. These associations are themselves modulated by
seasonal and interannual events in the El Niño–Southern Oscillation in winds and sea surface temperatures over the
tropical eastern Pacific Ocean. Efforts have been made to identify a possible
pathogen in air-filters flown at altitude above Japan. One source has been suggested in northeastern
China.
Genetics

Genetic susceptibility is suggested by increased
incidence among children of Japanese descent around the world, and also among
close and extended family members of affected people. Genetic factors are
also thought to influence development of coronary artery aneurysms and response
to treatment. The exact genetic contribution remains unknown. Genome-wide association studies and studies of individual candidate genes have together helped identify specific single nucleotide polymorphisms (SNPs), mostly found in genes with immune
regulatory functions. The associated genes and their levels of expression
appear to vary among different ethnic groups, both with Asian and non-Asian
backgrounds.
SNPs in FCGR2A, CASP3, BLK, ITPKC, CD40 and ORAI1 have all been linked to susceptibility, prognosis,
and risk of developing coronary artery aneurysms.
Various other
possible susceptibility genes have been proposed, including polymorphisms in the HLA region, but their significance is disputed.
Genetic
susceptibility to Kawasaki disease appears complex.
Gene–gene interactions also seem to affect susceptibility and prognosis.
At an epigenetic level, altered DNA methylation has been proposed as an early mechanistic factor
during the acute phase of the disease.
Diagnosis
|
Criteria for diagnosis |
|
Fever of ≥5 days' duration associated with at least
four† of these five changes |
|
Bilateral nonsuppurative conjunctivitis |
|
One or more changes of the mucous membranes of the upper respiratory tract, including throat redness, dry cracked lips, red
lips, and "strawberry"
tongue |
|
One or more changes of the arms and legs, including
redness, swelling, skin peeling around
the nails, and generalized peeling |
|
Polymorphous rash, primarily truncal |
|
Large lymph nodes in the neck (>15 mm in size) |
|
Disease cannot be explained by some other known
disease process |
|
†A diagnosis of Kawasaki disease can be made if
fever and only three changes are present if coronary artery disease is
documented by two-dimensional echocardiography or coronary angiography. |
|
Source: Nelson's essentials of
pediatrics, Review. |

Angiography showing ectatic LAD, with largest aneurysm = 6.5 mm in diameter
Since no specific laboratory test exists for Kawasaki
disease, diagnosis must be based on clinical signs and symptoms, together with laboratory findings. Timely diagnosis
requires careful history-taking and thorough physical examination. Establishing the diagnosis is difficult, especially
early in the course of the illness, and frequently children are not diagnosed
until they have seen several health-care providers. Many other serious
illnesses can cause similar symptoms, and must be considered in the
differential diagnosis, including scarlet fever, toxic shock syndrome, juvenile idiopathic arthritis, and childhood mercury poisoning (infantile acrodynia).
Classically, five days of feverplus of five diagnostic criteria must be met to establish the diagnosis.
The criteria are:
1.
erythema of the lips or oral cavity or cracking of the lips
2.
rash on the trunk
3.
swelling or erythema of the hands or feet
4.
red eyes (conjunctival injection)
5.
swollen lymph node in the neck of at least 15 mm
Many children, especially infants, eventually diagnosed
with Kawasaki disease, do not exhibit all of the above criteria. In fact, many
experts now recommend treating for Kawasaki disease even if only three days of
fever have passed and at least three diagnostic criteria are present,
especially if other tests reveal abnormalities consistent with Kawasaki
disease. In addition, the diagnosis can be made purely by the detection of
coronary artery aneurysms in the proper clinical setting.
Investigations
A physical examination will demonstrate many of the
features listed above.
Blood tests
·
Complete blood count may reveal normocytic anemia and eventually thrombocytosis.
·
Erythrocyte sedimentation rate will be elevated.
·
C-reactive
protein will be elevated.
·
Liver function tests may show evidence of hepatic inflammation and
low serum albumin levels.
Other optional tests include:
·
Electrocardiogram may show evidence of ventricular dysfunction or, occasionally, arrhythmia due to myocarditis.
·
Echocardiogram may show subtle coronary artery changes or, later, true
aneurysms.
·
Urinalysis may show white blood cells and protein in the urine
(pyuria and proteinuria) without evidence of bacterial growth.
·
Lumbar
punctureU
ltrasound or computerized tomography may show hydrops (enlargement) of the gallbladder.
·
may show evidence of aseptic
meningitis.
·
Angiography was historically used to detect coronary artery
aneurysms, and remains the gold standard for their detection, but is rarely
used today unless coronary artery aneurysms have already been detected by
echocardiography.
Biopsy is rarely performed, as it is not necessary for
diagnosis.

Subtypes
Based on clinical findings, a diagnostic distinction may be made between the 'classic' / 'typical' presentation of Kawasaki disease and 'incomplete' / 'atypical' presentation of a "suspected" form of the disease.[6] Regarding 'incomplete' / 'atypical' presentation, American Heart Association guidelines state that Kawasaki disease "should be considered in the differential diagnosis of prolonged unexplained fever in childhood associated with any of the principal clinical features of the disease, and the diagnosis can be considered confirmed when coronary artery aneurysms are identified in such patients by echocardiography.
A further distinction between 'incomplete' and 'atypical' subtypes may also be made in the presence of non-typical symptoms.
Case definition
For study purposes, including vaccine safety monitoring, an international case definition has been proposed to categorize 'definite' (i.e. complete/incomplete), 'probable' and 'possible' cases of Kawasaki disease
Differential
diagnosis
The broadness of the differential diagnosis is a challenge to timely diagnosis of Kawasaki disease.Infectious and noninfectious conditions requiring consideration include: measles and other viral infections (e.g. adenovirus, enterovirus); staphylococcal and streptococcal toxin-mediated diseases such as scarlet fever and toxic shock syndrome; drug hypersensitivity reactions (including Stevens Johnson syndrome); systemic onset juvenile idiopathic arthritis; Rocky Mountain spotted fever or other rickettsial infections; and leptospirosis. Infectious conditions that can mimic Kawasaki disease include periorbital cellulitis, peritonsillar abscess, retropharyngeal abscess, cervical lymphadenitis, parvovirus B19, mononucleosis, rheumatic fever, meningitis, staphylococcal scalded skin syndrome, toxic epidermal necrolysis, and Lyme disease.
Kawasaki-like
disease temporally associated with COVID-19.
In 2020, reports of a Kawasaki-like disease following exposure to SARS-CoV-2, the virus responsible for COVID-19, emerged in the US and Europe. The World Health Organization is examining possible links with COVID-19.] This emerging condition was named 'paediatric
multisystem inflammatory syndrome' by the Royal College of Paediatrics and Child Health, and 'multisystem inflammatory syndrome in children' by
the Centers for
Disease Control and Prevention. Guidance for diagnosis and reporting of cases has been
issued by these organizations.
Classification
Debate has occurred about whether Kawasaki disease should
be viewed as a characteristic immune response to some infectious pathogen, as an process, or as an autoinflammatory disease (i.e. involving innate rather than adaptive immune pathways). Overall, immunological research suggests that Kawasaki disease is
associated with a response to a conventional antigen (rather than a
superantigen) that involves both activation of the innate immune system and
also features of an adaptive immune response. Identification of the exact
nature of the immune process involved in Kawasaki disease could help guide
research aimed at improving clinical management.
Inflammation, or vasculitis, of the arteries and veins occurs throughout the body, usually caused by increased production of the cells of the immune system to a pathogen, or autoimmunity. Systemic vasculitides may be classified according to the type of cells involved in the proliferation, as well as the specific type of tissue damage occurring within the vein or arterial walls.[128] Under this classification scheme for systemic vasculitis, Kawasaki disease is considered to be a necrotizing vasculitis (also called necrotizing angiitis), which may be identified histologically by the occurrence of necrosis (tissue death), fibrosis, and proliferation of cells associated with inflammation in the inner layer of the vascular wall.
Other diseases involving necrotizing vasculitis
include polyarteritis nodosa, granulomatosis with polyangiitis, Henoch–Schönlein purpura, and eosinophilic granulomatosis with polyangiitis.
Kawasaki disease may be further classified as a medium-sized vessel vasculitis, affecting medium- and small-sized blood vessels, such as the smaller cutaneous vasculature (veins and arteries in the skin) that range from 50 to 100 µm diameter. Kawasaki disease is also considered to be a primary childhood vasculitis, a disorder associated with vasculitis that mainly affects children under the age of 18. A recent, consensus-based evaluation of vasculitides occurring primarily in children resulted in a classification scheme for these disorders, to distinguish them and suggest a more concrete set of diagnostic criteria for each. Within this classification of childhood vasculitides, Kawasaki disease is, again, a predominantly medium-sized vessel vasculitis.
It can also be classed as an autoimmune form of
vasculitis.
It is not associated with anti-neutrophil cytoplasmic antibodies, unlike other vasculitic disorders associated with them
such as granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis. This form of categorization is relevant for appropriate
treatment.
Treatment
Children with Kawasaki disease should be hospitalized and
cared for by a physician who has experience with this disease. In an academic
medical center, care is often shared between pediatric cardiology, pediatric rheumatology, and pediatric infectious
disease specialists (although no specific infectious agent has yet been
identified). To prevent damage to coronary arteries, treatment should be
started immediately following the diagnosis.
Intravenous immunoglobulin (IVIG) is the standard treatment for Kawasaki
disease]. and is
administered in high doses with marked improvement usually noted within 24
hours. If the fever does not respond, an additional dose may be considered. In
rare cases, a third dose may be given. IVIG is most useful within the first
seven days of fever onset, to prevent coronary artery aneurysm. IVIG given
within the first 10 days of the disease reduces the risk of damage to the
coronary arteries in children, without serious adverse effects.
Salicylate therapy, particularly aspirin, remains an important
part of the treatment (though questioned by some)[138] but salicylates alone are not as effective as IVIG.
There is limited evidence to indicate whether children should continue to
receive salicylate as part of their treatment. Aspirin therapy is started at high doses until the
fever subsides, and then is continued at a low dose when the patient returns
home, usually for two months to prevent blood clots from forming. Except for
Kawasaki disease and a few other indications, aspirin is otherwise normally not
recommended for children due to its association with Reye syndrome. Because children with Kawasaki disease will be taking
aspirin for up to several months, vaccination against varicella and influenza is required, as these infections are most likely to cause Reye
syndrome.
High-dose aspirin is associated with anemia and does not confer benefit to disease outcomes.
About 15-20% of children following the initial IVIG
infusion show persistent or recurrent fever and are classified as IVIG-resistant.
While the use of TNF alpha
blockers (TNF-α) may reduce treatment resistance and the infusion
reaction after treatment initiation, further research is needed.
Due to the potential involvement
of the upregulated calcium-nuclear factor of activated T cells pathway in the
development of the disease, a 2019 study found that the combination of
ciclosporin and IVIG infusion can suppress coronary artery abnormalities.
Further research is needed to determine which patients would respond best to
this treatment.
Corticosteroids have also been used, especially when other treatments fail or symptoms
recur, but in a randomized controlled trial, the addition of corticosteroid to
immune globulin and aspirin did not improve outcome. Additionally, corticosteroid use in the setting of
Kawasaki disease is associated with increased risk of coronary artery aneurysm,
so its use is generally contraindicated in this setting. In cases of Kawasaki
disease refractory to IVIG, cyclophosphamide and plasma exchange have been investigated as possible treatments, with
variable outcomes. However, a Cochrane review published in 2017 (updated in
2022) found that, in children, the use of corticosteroids in the acute phase of
KD was associated with improved coronary artery abnormalities, shorter hospital
stays, a decreased duration of clinical symptoms, and reduced inflammatory
marker levels. Patient populations based in Asia, people with higher risk
scores, and those receiving longer steroid treatment may have greater benefit
from steroid use.
Prognosis
With early treatment, rapid recovery from the acute
symptoms can be expected, and the risk of coronary artery aneurysms is greatly
reduced. Untreated, the acute symptoms of Kawasaki disease are self-limited (i.e. the
patient will recover eventually), but the risk of coronary artery involvement
is much greater, even many years later. Many cases of myocardial infarction in
young adults have now been attributed to Kawasaki disease that went undiagnosed
during childhood. Overall, about 2% of patients die from
complications of coronary vasculitis.
Laboratory evidence of increased inflammation combined
with demographic features (male sex, age less than six months or greater than
eight years) and incomplete response to IVIG therapy create a profile of a
high-risk patient with Kawasaki disease. The likelihood that an aneurysm will resolve
appears to be determined in large measure by its initial size, in which the
smaller aneurysms have a greater likelihood of regression Other factors are
positively associated with the regression of aneurysms, including being younger
than a year old at the onset of Kawasaki disease, fusiform rather than sacc This severe outcome may
require further treatment such as percutaneous transluminal angioplasty coronary artery stenting, bypass grafting, and even cardiac transplantation.
A relapse of symptoms may occur soon after initial treatment
with IVIG. This usually requires rehospitalization and retreatment. Treatment
with IVIG can cause allergic and nonallergic acute reactions, aseptic
meningitis, fluid overload, and rarely, other serious reactions. Overall,
life-threatening complications resulting from therapy for Kawasaki disease are
exceedingly rare, especially compared with the risk of nontreatment. Evidence
indicates Kawasaki disease produces altered lipid metabolism that persists beyond the clinical resolution of the
disease.
Rarely, recurrence can occur in Kawasaki disease with or without treatment.
Epidemiology
Kawasaki disease affects boys more than girls, with
people of Asian ethnicity, particularly Japanese people. The higher incidence
in Asian populations is thought to be linked to genetic susceptibility. Incidence rates vary between countries.
Currently, Kawasaki disease is the most commonly diagnosed pediatric vasculitis in the world. By far, the highest incidence of Kawasaki disease occurs in Japan, with the most recent study placing the attack rate at 218.6 per 100,000 children less than five years of age (about one in 450 children). At this present attack rate, more than one in 150 children in Japan will develop Kawasaki disease during their lifetimes.
However, its incidence in the United States is
increasing. Kawasaki disease is predominantly a disease of young children, with
80% of patients younger than five years of age. About 2,000–4,000 cases are
identified in the U.S. each year (9 to 19 per 100,000 children younger than
five years of age). In the continental United States, Kawasaki disease
is more common during the winter and early spring, boys with the disease
outnumber girls by ≈1.5–1.7:1, and 76% of affected children are less than 5
years of age.
In the United Kingdom, prior to 2000, it was diagnosed in fewer than one in every 25,000 people per year. Incidence of the disease doubled from 1991 to 2000, however, with four cases per 100,000 children in 1991 compared with a rise of eight cases per 100,000 in 2000.[162] By 2017, this figure had risen to 12 in 100,000 people with 419 diagnosed cases of Kawasaki disease in the United Kingdom.
In Japan, the rate is 240 in every 100,000 people.
Coronary artery aneurysms due to Kawasaki disease are
believed to account for 5% of acute coronary syndrome cases in adults under 40
years of age.
History
The disease was first reported by Tomisaku Kawasaki in a
four-year-old child with a rash and fever at the Red Cross Hospital in Tokyo in
January 1961, and he later published a report on 50 similar cases. Later, Kawasaki and colleagues were persuaded of
definite cardiac involvement when they studied and reported 23 cases, of which
11 (48%) patients had abnormalities detected by an electrocardiogram. In 1974, the first description of this disorder was
published in the English-language literature. In 1976, Melish et al. described the same illness
in 16 children in Hawaii. Melish and Kawasaki had independently developed the
same diagnostic criteria for the disorder, which are still used today to make
the diagnosis of classic Kawasaki disease. Dr. Kawasaki died on June 5, 2020,
at the age of 95.
A question was raised whether the disease only started
during the period between 1960 and 1970, but later a preserved heart of a
seven-year-old boy who died in 1870 was examined and showed three aneurysms of
the coronary arteries with clots, as well as pathologic changes consistent with
Kawasaki disease. Kawasaki disease is now recognized worldwide. Why
cases began to emerge across all continents around the 1960s and 1970s is
unclear. Possible explanations could include confusion with
other diseases such as scarlet fever, and easier recognition stemming from
modern healthcare factors such as the widespread use of antibiotics. In particular, old pathological descriptions from
Western countries of infantile polyarteritis nodosa coincide with reports of
fatal cases of Kawasaki disease.
In the United States and other developed nations, Kawasaki disease appears to have replaced acute rheumatic fever as the most common cause of acquired heart disease in children.
Jan Ricks Jennings, MHA , LFACHE
Senior Consultant
Senior Management Resources, LLC
JanJenningsBlog.Blogspot.com
412.9130636 Cell
724.733.0509 Office
February 28, 2023
This
article was published on February 28th. On February 28, 1906 the notorious American Gangster,
Benjamin "Bugsy" Siegel, was born.. He was an
American mobster who
was a driving force behind the development of the Las Vegas Strip. Siegel was not only influential within
the Jewish Mob,
but along with his childhood friend and fellow gangster Meyer Lansky,
also held significant influence within the Italian-American Mafia and
the largely Italian-Jewish National Crime Syndicate. Described as
handsome and charismatic, he became one of the first front-page celebrity
gangsters.
Siegel
was one of the founders and leaders of Murder,
Inc. and became a bootlegger during Prohibition.
After the Twenty-first Amendment was
passed repealing Prohibition in 1933, he turned to gambling.
In 1936, he left New York and
moved to California. His
time as a mobster during this period was mainly as a hitman and
muscle, as he was noted for his prowess with guns and violence. In 1941, Siegel
was tried for the murder of friend and fellow mobster Harry Greenberg,
who had turned informant. He was acquitted in
1942.
Siegel
traveled to Las Vegas, Nevada,
where he handled and financed some of the original casinos.
He assisted developer William
R. Wilkerson's Flamingo
Hotel after Wilkerson ran out of funds.
Siegel assumed control of the
project and managed the final stages of construction. The Flamingo opened on
December 26, 1946, in a driving rainstorm, resulting in a poor reception and
technical difficulties, and soon closed. It reopened in March 1947 with a
finished hotel, but by then his mob partners were convinced an estimated $1
million of the construction budget overage had been skimmed by either
girlfriend Virginia
Hill, Siegel or both. On June 20, 1947,
Siegel was shot dead by a sniper through the window of Hill's Linden Drive
mansion in Beverly Hills, California. He died at age 41.
No comments:
Post a Comment